









Table des matières
Sclérose tubéreuse de Bourneville
Qu'Est-ce que c'est ?
La sclérose tubéreuse de Bourneville est une maladie génétique complexe caractérisée par le développement d'une tumeur bénigne (non cancéreuse) dans différentes parties du corps. Ces tumeurs peuvent alors être localisées dans la peau, le cerveau, les reins et d'autres organes et tissus. Cette pathologie peut également causer de graves problèmes dans le développement de l'individu. Cependant, les manifestations cliniques et la gravité de la maladie varient d'un patient à l'autre.
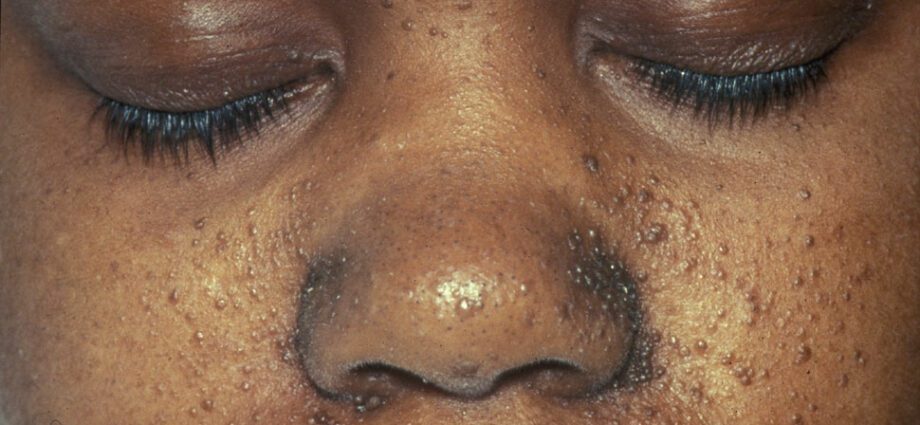
Les anomalies cutanées associées sont généralement similaires à des taches sur la peau ou à des zones où la peau est plus claire que sur le reste du corps. Le développement de tumeurs au visage est appelé angiofibrome.
Dans le cadre de lésions cérébrales, les signes cliniques sont des crises d'épilepsie, des troubles du comportement (hyperactivité, agressivité, déficience intellectuelle, troubles des apprentissages, etc.). Certains enfants atteints de la maladie souffrent même d'une forme d'autisme, de troubles du développement, affectant les interactions sociales et la communication. Les tumeurs cérébrales bénignes peuvent également entraîner des complications qui peuvent être mortelles pour le sujet.
Le développement de tumeurs dans les reins est fréquent chez les personnes atteintes de sclérose tubéreuse de Bourneville. Cela peut entraîner de graves complications dans la fonction rénale. De plus, des tumeurs peuvent se développer dans le cœur, les poumons et la rétine. (2)
C'est une maladie rare, dont la prévalence (nombre de cas dans une population donnée à un moment donné) s'élève à 1/8 à 000/1 personne. (15)
Symptômes
Les manifestations cliniques associées à la sclérose tubéreuse de Bourneville varient selon les organes atteints. De plus, les symptômes associés à la maladie varient considérablement d'un individu à l'autre. Avec des symptômes allant de légers à graves.
Les symptômes les plus largement identifiés de cette maladie sont les crises d'épilepsie, les troubles cognitifs et comportementaux, les anomalies cutanées, etc. Les organes les plus souvent touchés sont : le cerveau, le cœur, les reins, les poumons et la peau.
Le développement de tumeurs malignes (cancéreuses) est possible dans cette maladie mais sont rares et touchent principalement les reins.
Les signes cliniques de la maladie dans le cerveau proviennent d'attaques à différents niveaux :
– atteinte des tubercules corticaux ;
– nodules épendymaires (SEN) ;
– les astrocytomes épendymaires géants.
Ils se traduisent par : le développement d'un retard mental, de difficultés d'apprentissage, de troubles du comportement, d'agressivité, de troubles de l'attention, d'hyperactivité, de troubles obsessionnels compulsifs, etc.
Les lésions rénales se caractérisent par le développement de kystes ou d'angiomyolipomes. Ceux-ci peuvent entraîner des douleurs rénales et même une insuffisance rénale. Si des saignements abondants sont perceptibles, cela peut être dû à une anémie sévère ou à une pression artérielle élevée. D'autres conséquences plus graves mais rares peuvent également être visibles, notamment le développement de carcinomes (tumeur des cellules constitutives de l'épithélium).
Les lésions oculaires peuvent être similaires à des taches visibles sur la rétine, provoquant des troubles visuels ou même la cécité.
Les anomalies cutanées sont nombreuses :
– les macules hypomélaniques : qui se traduisent par l'apparition de taches claires sur la peau, n'importe où sur le corps, dues à une carence en mélanine, une protéine qui donne de la couleur à la peau ;
– l'apparition de taches rouges sur le visage ;
– des plaques décolorées sur le front ;
– d'autres anomalies cutanées, dépendantes d'un individu à l'autre.
Des lésions pulmonaires sont présentes chez 1/3 des patients avec une légère prédominance féminine. Les symptômes associés sont alors des difficultés respiratoires plus ou moins sévères.
Les origines de la maladie
L'origine de la maladie est génétique et héréditaire.
La transmission implique des mutations dans les gènes TSC1 et TSC2. Ces gènes d'intérêt entrent en jeu dans la formation de protéines : l'hamartine et la tubérine. Ces deux protéines permettent, à travers un jeu interactif, de réguler la prolifération cellulaire.
Les patients atteints de la maladie naissent avec au moins une copie mutée de ces gènes dans chacune de leurs cellules. Ces mutations limitent alors la formation d'hamartine ou de tuberculine.
Dans le contexte où les deux copies du gène sont mutées, elles empêchent totalement la production de ces deux protéines. Cette carence en protéines ne permet donc plus à l'organisme de réguler la croissance de certaines cellules et, en ce sens, conduit au développement de cellules tumorales dans différents tissus et/ou organes.
Les facteurs de risque
Les facteurs de risque de développer une telle pathologie sont génétiques.
En effet, la transmission de la maladie est efficace selon un mode autosomique dominant. Soit, le gène muté d'intérêt est situé sur un chromosome non sexuel. De plus, la présence d'une seule des deux copies du gène muté est suffisante pour que la maladie se développe.
En ce sens, un individu possédant l'un de ces deux parents atteint de la maladie a 50% de risque de développer lui-même le phénotype malade.
Prévention et traitement
Le diagnostic de la maladie est d'abord différentiel. Elle repose sur des critères physiques atypiques. Dans la plupart des cas, les premiers signes caractéristiques de la maladie sont : la présence de crises d'épilepsie récurrentes et des retards de développement du sujet. Dans d'autres cas, ces premiers signes se traduisent par des taches cutanées ou l'identification d'une tumeur cardiaque.
Suite à ce premier diagnostic, des examens complémentaires sont indispensables afin de valider ou non le diagnostic. Ceux-ci inclus:
– un scanner cérébral ;
– une IRM (Imagerie par Résonance Magnétique) du cerveau ;
– une échographie du cœur, du foie et des reins.
Le diagnostic peut être effectif à la naissance de l'enfant. Dans le cas contraire, il est important qu'elle soit réalisée le plus rapidement possible afin de prendre en charge le patient le plus rapidement possible.
Actuellement, il n'y a pas de remède pour la maladie. Les traitements associés sont donc indépendants des symptômes présentés par chaque individu.
Habituellement, des médicaments antiépileptiques sont administrés pour limiter les crises. En outre, des médicaments pour le traitement des cellules tumorales du cerveau et des reins sont également prescrits. Dans le cadre de troubles du comportement, une prise en charge spécifique de l'enfant est nécessaire.
Le traitement de la maladie est généralement à long terme. (1)