









Table des matières
Le sarcome d'Ewing
Qu'Est-ce que c'est ?
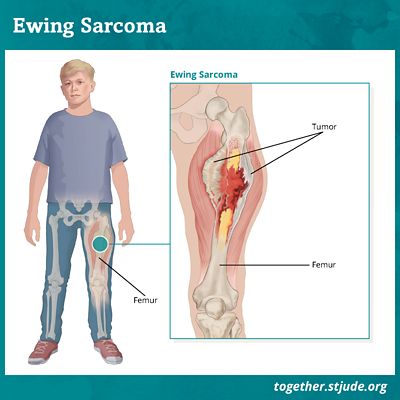
Le sarcome d'Ewing est caractérisé par le développement d'une tumeur maligne dans les os et les tissus mous. Cette tumeur a la particularité d'être à fort potentiel métastatique. Soit la propagation des cellules tumorales dans tout l'organisme est souvent identifiée dans cette pathologie.
C'est une maladie rare touchant plus généralement les enfants. Son incidence s'élève à 1/312 enfants de moins de 500 ans.
La tranche d'âge la plus touchée par le développement de cette forme tumorale se situe entre 5 et 30 ans, avec une incidence encore plus importante entre 12 et 18 ans. (3)
Les manifestations cliniques associées sont une douleur et un gonflement à l'emplacement de la tumeur.
Les localisations des cellules tumorales caractéristiques du sarcome d'Ewing sont multiples : jambes, bras, pieds, mains, poitrine, bassin, crâne, colonne vertébrale, etc.
Ce sarcome d'Ewing est aussi appelé : tumeur neuroectodermique périphérique primitive. (1)
Les examens médicaux permettent le diagnostic éventuel de la maladie et déterminent son stade d'évolution. L'examen le plus souvent associé est une biopsie.
Des facteurs et des conditions spécifiques peuvent avoir un impact sur le pronostic de la maladie chez un sujet affecté. (1)
Ces facteurs incluent notamment la propagation des cellules tumorales aux seuls poumons, dont le pronostic est plus favorable, ou le développement de formes métastatiques vers d'autres parties du corps. Dans ce dernier cas, le pronostic est plus sombre.
De plus, la taille de la tumeur et l'âge de la personne atteinte ont un rôle fondamental dans le pronostic vital. En effet, dans le cas où la taille de la tumeur s'élève à plus de 8 cm, le pronostic est plus préoccupant. Quant à l'âge, plus le diagnostic de la pathologie est posé tôt, meilleur est le pronostic pour le patient. (4)
Le sarcome d'Ewing est l'un des trois principaux types de cancer primitif des os avec le chondrosarcome et l'ostéosarcome. (2)
Symptômes
Les symptômes les plus couramment associés au sarcome d'Ewing sont une douleur visible et un gonflement des os et des tissus mous touchés.
Les manifestations cliniques suivantes peuvent provenir du développement d'un tel sarcome : (1)
- douleur et/ou gonflement dans les bras, les jambes, la poitrine, le dos ou le bassin ;
- la présence de « bosses » sur ces mêmes parties du corps ;
- la présence de fièvre sans raison particulière ;
- fractures osseuses sans raison sous-jacente.
Les symptômes associés dépendent néanmoins de la localisation de la tumeur ainsi que de son importance en termes de développement.
La douleur ressentie par le patient atteint de cette pathologie s'intensifie généralement avec le temps.
D'autres symptômes moins courants peuvent également être visibles, tels que : (2)
- une fièvre élevée et persistante;
- raideur musculaire;
- perte de poids importante.
Cependant, un patient atteint du sarcome d'Ewing peut ne présenter aucun symptôme. En ce sens, la tumeur peut alors se développer sans manifestation clinique particulière et ainsi toucher l'os ou les tissus mous sans être visible. Le risque de fracture est d'autant plus important dans ce dernier cas. (2)
Les origines de la maladie
Comme le sarcome d'Ewing est une forme de cancer, on sait peu de choses sur les origines exactes de son développement.
Une hypothèse a néanmoins été avancée concernant la cause de son développement. En effet, le sarcome d'Ewing touche particulièrement les enfants de plus de 5 ans et les adolescents. En ce sens, la possibilité d'un lien entre la croissance osseuse rapide de cette catégorie de personnes et le développement du sarcome d'Ewing a été évoquée.
La période de puberté chez les enfants et les adolescents rend les os et les tissus mous plus vulnérables au développement d'une tumeur.
La recherche a également montré qu'un enfant né avec une hernie ombilicale est trois fois plus susceptible de développer le sarcome d'Ewing. (2)
Au-delà de ces hypothèses évoquées ci-dessus, l'origine de la présence d'une translocation génétique a également été avancée. Cette translocation implique le gène EWSRI (22q12.2). Une translocation t (11 ; 22) (q24 ; q12) au sein de ce gène d'intérêt a été trouvée dans près de 90 % des tumeurs. De plus, de nombreuses variantes génétiques ont fait l'objet d'investigations scientifiques, impliquant les gènes ERG, ETV1, FLI1 et NR4A3. (3)
Les facteurs de risque
Du point de vue où les origines exactes de la pathologie sont, à ce jour, encore mal connues, les facteurs de risque le sont aussi.
De plus, selon les résultats d'études scientifiques, un enfant né avec une hernie ombilicale serait trois fois plus susceptible de développer un type de cancer.
De plus, au niveau génétique, la présence de translocations au sein du gène EWSRI (22q12.2) ou de variants génétiques dans les gènes ERG, ETV1, FLI1 et NR4A3, peut faire l'objet de facteurs de risque supplémentaires de développer la maladie. .
Prévention et traitement
Le diagnostic du sarcome d'Ewing repose sur un diagnostic différentiel par la présence de symptômes caractéristiques chez le patient.
Suite à l'analyse par le médecin des zones douloureuses et enflées, une radiographie est généralement prescrite. D'autres systèmes d'imagerie médicale peuvent également être utilisés, tels que : l'imagerie par raisonnement magnétique (IRM) ou encore les scans.
Une biopsie osseuse peut également être réalisée pour confirmer ou non le diagnostic. Pour cela, un échantillon de moelle osseuse est prélevé et analysé au microscope. Cette technique de diagnostic peut être réalisée après une anesthésie générale ou locale.
Le diagnostic de la maladie doit être réalisé le plus tôt possible afin que la prise en charge se fasse rapidement et ainsi le pronostic soit meilleur.
Le traitement du sarcome d'Ewing est similaire au traitement général des autres cancers : (2)
- la chirurgie est un moyen efficace de traiter ce type de sarcome. Cependant, l'intervention chirurgicale dépend de la taille de la tumeur, de sa localisation ainsi que de son niveau de propagation. Le but de la chirurgie est de remplacer la partie de l'os ou des tissus mous endommagée par la tumeur. Pour cela, une prothèse métallique ou une greffe osseuse peut être utilisée dans le remplacement de la zone touchée. Dans les cas extrêmes, l'amputation d'un membre est parfois nécessaire pour prévenir la rechute du cancer ;
- chimiothérapie, généralement utilisée après la chirurgie pour rétrécir la tumeur et faciliter la cicatrisation.
- la radiothérapie, est également souvent utilisée après une chimiothérapie, avant ou après une intervention chirurgicale pour réduire la taille de la tumeur et éviter le risque de rechute.