









Table des matières
maladie de Hirschsprung
Qu'Est-ce que c'est ?
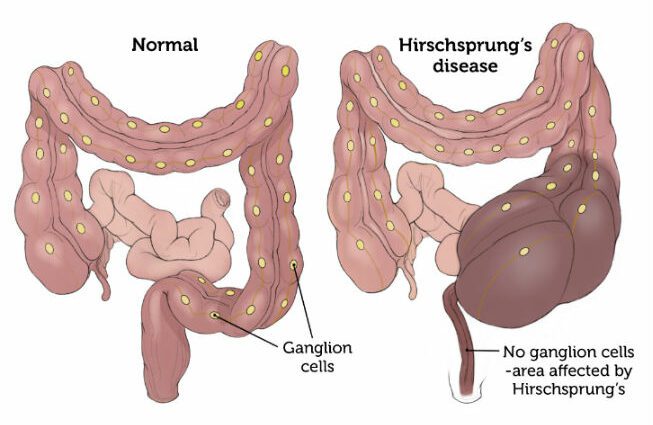
La maladie de Hirschsprung (HSCR) est caractérisée par une paralysie de la partie terminale du gros intestin.
Cette pathologie apparaît dès la naissance et est la conséquence d'une absence de ganglions nerveux (cellules formant un renflement sur le trajet du nerf) dans la paroi de l'intestin.
La déglutition des aliments par le tube digestif jusqu'à leur élimination est en grande partie possible grâce au péristaltisme intestinal. Ce péristaltisme est un ensemble de contractions des muscles intestinaux permettant la progression du bol alimentaire le long du tube digestif.
Dans cette situation où il y a absence de ganglions nerveux dans le gros intestin, le péristaltisme n'est plus assuré par l'organisme. En ce sens, une dilatation de l'intestin et une augmentation de son volume sont créées.
Les symptômes associés sont d'autant plus importants que la zone des ganglions nerveux est importante. (1)
Cette maladie se définit donc par des symptômes intestinaux atypiques : occlusion intestinale. C'est un blocage du transit et des gaz entraînant des douleurs abdominales, des coliques (crampes intestinales), des nausées, des ballonnements, etc.
La HSCR affecte environ 1 naissance sur 5 par an. La forme affectant la partie terminale du côlon (gros intestin) touche principalement les garçons. (000) Les filles sont plus sujettes au développement de cette maladie sous une forme plus répandue. (2)
Cette pathologie touche majoritairement les bébés et les jeunes enfants. (3)
Plusieurs formes de la maladie ont été mises en évidence (2) :
– la forme « classique », ou encore appelée « forme segment court ». Cette forme est la plus fréquente chez les patients atteints de cette pathologie, jusqu'à 80%. Cette forme de la maladie affecte la partie terminale du côlon jusqu'au segment rectal ;
– la forme « segment long », qui s'étend au côlon sigmoïde, touche près de 15 % des patients ;
– la forme « colique totale », affectant l'ensemble du côlon, concerne 5 % des patients.
Symptômes
Le transit intestinal est contrôlé par le système nerveux. Les ganglions nerveux sont donc localisés dans l'intestin permettant le transfert d'informations du cerveau pour le contrôle du péristaltisme intestinal et donc la progression des aliments le long du tube digestif.
Une absence de ces ganglions, dans le cas de la maladie de Hirschsprung, empêche toute transmission d'informations et bloque ainsi le péristaltisme intestinal. Les aliments ne peuvent plus passer par les intestins et se retrouvent bloqués dans le tube digestif.
Les symptômes de cette maladie sont généralement perceptibles très tôt à la naissance. Cependant, dans certains cas, ils peuvent apparaître après un ou deux ans. (3)
Les symptômes affectant les nouveau-nés et les enfants sont principalement :
– difficultés de transit ;
– une incapacité à expulser le méconium (premiers excréments du nouveau-né) pendant les 48 premières heures ;
- constipation;
– la jaunisse ;
– vomissements ;
- la diarrhée;
- douleur abdominale;
– la sous-alimentation.
Les symptômes affectant les enfants plus âgés sont :
– constipation sévère avec complications (retard de croissance en taille et en poids) ;
– une mauvaise alimentation ;
- distension de l'abdomen;
- une fièvre.
L'enfant peut également développer des infections intestinales, telles que l'entérocolite.
Des anomalies supplémentaires peuvent également être visibles : surdité de perception (syndrome de Waardenburg-Shah), déficience intellectuelle (syndrome de Mowat-Wilson), hypoventilation alvéolaire centrale (syndrome de Haddad), anomalies des membres (syndrome de Bardet) Biedl), cancer médullaire de la thyroïde (syndrome endocrinien multiple). néoplasie de type 2B) ou des anomalies chromosomiques (syndrome de Down). (2)
Les origines de la maladie
La maladie de Hirschsprung est causée par une anomalie dans le développement du système nerveux entérique. Il s'agit d'une aganglionose, c'est-à-dire d'une absence de ganglions nerveux (appelés aussi « cellules de Cajal ») dans les intestins. Ce déficit ganglionnaire est plus particulièrement localisé dans la partie terminale du gros intestin (côlon).
Chez le sujet atteint de cette pathologie, cette partie de l'intestin reste donc dans un état de contraction tonique et permanente. Cette situation conduit à une occlusion intestinale. (2)
Des facteurs génétiques et environnementaux ont été impliqués dans le développement de la maladie de Hirschsprung. (2)
En effet, certains gènes ont été mis en évidence dans le développement de cette pathogenèse. C'est une maladie polygénétique qui concerne notamment les gènes :
- Proco-oncogène ret (RET) ;
– le gène du facteur neutrotrophique dérivé des cellules gliales (GDNF) ;
– le gène du récepteur de l'endothéline de type B (EDNRB) ;
– le gène de l'endothéline 3 (EDN3) ;
– le gène de l'enzyme de conversion 1 de l'endothéline 1 (ECE1) ;
– le gène de la molécule d'adhésion cellulaire L1 (L1CAM).
Les facteurs de risque
Comme indiqué précédemment, la maladie de Hirschsprung est la conséquence de l'absence de ganglions nerveux dans le gros intestin jusqu'à l'anus, empêchant le péristaltisme intestinal et donc la remontée des aliments à ce niveau.
Ce déficit en cellules de Cajal (ganglions nerveux) est la conséquence d'un déficit de croissance de ces cellules au cours du développement fœtal. Les causes de ce manque de croissance cellulaire avant la naissance ne sont pas encore connues. Néanmoins, la possibilité d'une relation entre l'état de santé général de la mère pendant sa période de grossesse et l'absence de ce type de cellules chez le fœtus a été avancée.
De nombreux gènes ont été mis en évidence dans le développement de la maladie. La présence de ces gènes peut être fréquente au sein d'une même famille. Une partie de l'hérédité serait alors à l'origine du développement de cette maladie.
Par ailleurs, certaines pathologies peuvent également être un facteur de risque supplémentaire en termes de développement de la maladie de Hirschsprung. C'est notamment le cas du syndrome de Down. (3)
Prévention et traitement
Le diagnostic différentiel est fait en fonction des symptômes caractéristiques de la maladie présentés par le sujet : occlusion intestinale, sténose ano-rectale, tumeurs pelviennes, etc. (2)
Le diagnostic le plus souvent associé à la maladie est posé par une biopsie rectale. Cette biopsie montre la présence ou l'absence de ganglions nerveux dans le gros intestin. A cela s'ajoute une surexpression de l'acétylcholine estérase (enzyme permettant d'hydrolyser l'acétylcholine en acide acétique et choline). (2)
Un lavement baryté (examen radiographique pour visualiser le gros intestin) peut également être réalisé dans le diagnostic de cette pathologie. Cette méthode permet de visualiser une zone transitoire d'absence de cellules nerveuses, indiquant le développement de la maladie d'Hischsprung. Cependant, cette technique de diagnostic n'est pas fiable à 100 %. En effet, 10 à 15 % des cas de maladie de Hirschsprung ne seraient pas diagnostiqués après cette tentative de diagnostic. (4)
Le traitement le plus important de la maladie est la chirurgie. Il permet l'ablation de la partie de l'intestin déficiente en cellules nerveuses. (4)
En cas de lésion totale du côlon, une greffe du côlon peut être nécessaire. (2)
Suite à cela, une stomie (technique chirurgicale permettant de faire la connexion entre deux organes) peut être réalisée afin de relier la partie opérée de l'intestin à l'anus ou à la partie supérieure de l'intestin. Cette stomie peut être permanente ou temporaire selon les cas. (4)
La chirurgie aide à réduire les symptômes associés à la maladie. Cependant, le pronostic n'est pas complet et des complications inflammatoires peuvent apparaître et être mortelles.