









Table des matières
Syndrome de Treacher-Collins
Maladie génétique rare, le syndrome de Teacher-Collins se caractérise par le développement de malformations congénitales du crâne et du visage au cours de la vie embryonnaire, entraînant des déformations du visage, des oreilles et des yeux. Les conséquences esthétiques et fonctionnelles sont plus ou moins sévères et certains cas nécessitent de nombreuses interventions chirurgicales. Cependant, dans la plupart des cas, la prise en charge permet de préserver une certaine qualité de vie.
Qu'est-ce que le syndrome de Treacher-Collins ?
Définition
Le syndrome de Treacher-Collins (du nom d'Edward Treacher Collins, qui l'a décrit pour la première fois en 1900) est une maladie congénitale rare qui se manifeste dès la naissance par des malformations plus ou moins sévères de la partie inférieure du corps. visage, yeux et oreilles. Les attaques sont bilatérales et symétriques.
Ce syndrome est également appelé syndrome de Franceschetti-Klein ou dysostose mandibulo-faciale sans anomalies terminales.
Causes
Trois gènes sont jusqu'à présent connus pour être impliqués dans ce syndrome :
- le gène TCOF1, situé sur le chromosome 5,
- les gènes POLR1C et POLR1D, situés respectivement sur les chromosomes 6 et 13.
Ces gènes dirigent la production de protéines qui jouent un rôle clé dans le développement embryonnaire des structures faciales. Leur altération par mutations perturbe le développement des structures osseuses (principalement celles des mâchoires inférieure et supérieure et des pommettes) et des tissus mous (muscles et peau) de la partie inférieure du visage au cours du deuxième mois de gestation. Le pavillon de l'oreille, le conduit auditif ainsi que les structures de l'oreille moyenne (osselets et/ou tympans) sont également touchés.
Diagnostique
Des malformations faciales peuvent être suspectées dès l'échographie du deuxième trimestre de la grossesse, notamment en cas de malformations importantes de l'oreille. Dans ce cas, le diagnostic prénatal sera établi par une équipe pluridisciplinaire à partir de l'imagerie par résonance magnétique (IRM) du fœtus, permettant de visualiser les malformations avec plus de précision.
La plupart du temps, le diagnostic est posé par un examen physique effectué à la naissance ou peu après. En raison de la grande variabilité des malformations, elle doit être confirmée dans un centre spécialisé. Un test génétique sur un échantillon de sang peut être ordonné pour rechercher les anomalies génétiques impliquées.
Certaines formes bénignes passent inaperçues ou seront détectées fortuitement tardivement, par exemple suite à l'apparition d'un nouveau cas dans la famille.
Une fois le diagnostic posé, l'enfant est soumis à une série d'examens complémentaires :
- imagerie faciale (radiographie, tomodensitométrie et IRM),
- examens des oreilles et tests auditifs,
- évaluation de la vue,
- recherche d’apnée du sommeil (polysomnographie)…
Les personnes concernées
On pense que le syndrome de Treacher-Collins affecte un nouveau-né sur 50, filles et garçons. On estime qu'environ 000 nouveaux cas apparaissent chaque année en France.
Les facteurs de risque
Un conseil génétique dans un centre de référence est recommandé pour évaluer les risques de transmission génétique.
Environ 60 % des cas apparaissent isolés : l'enfant est le premier patient de la famille. Les malformations surviennent à la suite d'un accident génétique ayant touché l'une ou l'autre des cellules reproductrices impliquées dans la fécondation (mutation « de novo »). Le gène muté sera alors transmis à sa descendance, mais il n'y a pas de risque particulier pour ses frères et sœurs. Cependant, il convient de vérifier si l'un de ses parents n'est pas réellement atteint d'une forme mineure du syndrome et porteur de la mutation sans le savoir.
Dans d'autres cas, la maladie est héréditaire. Le plus souvent, le risque de transmission est d'un sur deux à chaque grossesse, mais selon les mutations impliquées, il existe d'autres modes de transmission.
Symptômes du syndrome de Treacher-Collins
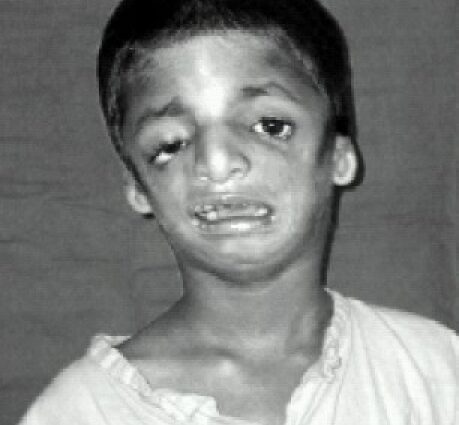
Les traits du visage des personnes atteintes sont souvent caractéristiques, avec un menton atrophié et fuyant, des pommettes inexistantes, des yeux inclinés vers le bas vers les tempes, des oreilles à pavillon petit et mal ourlé, voire totalement absents…
Les principaux symptômes sont liés à des malformations de la sphère ORL :
Difficultés respiratoires
De nombreux enfants naissent avec des voies respiratoires supérieures étroites et une bouche étroitement ouverte, avec une petite cavité buccale largement obstruée par la langue. D'où des difficultés respiratoires importantes notamment chez les nouveau-nés et les nourrissons, qui se traduisent par des ronflements, des apnées du sommeil et une respiration trop faible.
Difficulté à manger
Chez le nourrisson, l'allaitement peut être entravé par des difficultés respiratoires et par des anomalies du palais et du voile du palais, parfois dédoublés. L'alimentation est plus facile après l'introduction d'aliments solides, mais la mastication peut être difficile et les problèmes dentaires sont fréquents.
Surdité
Une gêne auditive due à des malformations de l'oreille externe ou moyenne est présente dans 30 à 50 % des cas.
Troubles visuels
Un tiers des enfants souffrent de strabisme. Certains peuvent également être myopes, hypermétropes ou astigmates.
Difficultés d'apprentissage et de communication
Le syndrome de Treacher-Collins n'entraîne pas de déficit intellectuel, mais la surdité, les troubles visuels, les difficultés d'élocution, les répercussions psychologiques de la maladie ainsi que les troubles induits par des soins médicaux souvent très lourds peuvent entraîner un retard. langage et difficulté à communiquer.
Traitements du syndrome de Treacher-Collins
Soins infantiles
Une assistance respiratoire et/ou une alimentation par sonde peuvent être nécessaires pour faciliter la respiration et l'alimentation du nourrisson, parfois dès la naissance. Lorsque l'assistance respiratoire doit être maintenue dans le temps, une trachéotomie (petite ouverture dans la trachée, au niveau du cou) est réalisée pour introduire une canule assurant directement le passage de l'air dans les voies respiratoires.
Traitement chirurgical des malformations
Des interventions chirurgicales plus ou moins complexes et nombreuses, portant sur le voile du palais, les mâchoires, le menton, les oreilles, les paupières et le nez peuvent être proposées pour faciliter l'alimentation, la respiration ou l'audition, mais aussi pour réduire l'impact esthétique des malformations.
A titre indicatif, les fentes du voile du palais sont fermées avant l'âge de 6 mois, les premiers gestes esthétiques sur les paupières et les pommettes à partir de 2 ans, l'allongement de la mandibule (distraction mandibulaire) vers 6 ou 7 ans, la remise en place de le pavillon de l'oreille vers 8 ans, élargissement des conduits auditifs et/ou chirurgie des osselets vers 10 à 12 ans… D'autres opérations de chirurgie esthétique peuvent encore être pratiquées à l'adolescence…
Dispositif d'écoute pour malentendant
Les appareils auditifs sont parfois possibles dès l'âge de 3 ou 4 mois lorsque la surdité touche les deux oreilles. Différents types de prothèses sont disponibles selon la nature des dommages, avec une bonne efficacité.
Suivi médical et paramédical
Afin de limiter et de prévenir le handicap, un suivi régulier est pluridisciplinaire et fait appel à divers spécialistes :
- ORL (risque élevé d'infection)
- Ophtalmologiste (correction des troubles visuels) et orthoptiste (rééducation oculaire)
- Dentiste et orthodontiste
- Orthophoniste…
Un accompagnement psychologique et pédagogique est souvent nécessaire.